The dystrophin is a rod or rod-shaped protein associated with the membrane of skeletal, smooth and cardiac muscle cells, also present in nerve cells and in other organs of the human body.
It has functions similar to those of other cytoskeletal proteins, and is believed to work primarily on muscle fiber membrane stability and binding of the extracellular basement membrane with the intracellular cytoskeleton..
It is encoded on the X chromosome, in one of the largest genes described for humans, some of whose mutations are involved in pathologies linked to the sex chromosomes, such as Duchenne muscular dystrophy (DMD).
This pathology is the second most common inherited disorder in the world. It affects one in every 3,500 men and it becomes evident between the ages of 3 and 5 as accelerated muscle wasting that can reduce life span to no more than 20 years.
The dystrophin gene was isolated for the first time in 1986 and was characterized using positional cloning, which meant a great advance for the molecular genetics of the time..
Article index
Dystrophin is a very diverse protein that is associated with the plasma membrane of muscle cells (sarcolemma) and with that of other cells in different body systems.
Its diversity is due to the processes that are related to the regulation of the expression of the gene that encodes it, which is one of the largest genes described for human beings. This is because it has more than 2.5 million base pairs, which represent about 0.1% of the genome..
This gene is predominantly expressed in skeletal and cardiac muscle cells and also in the brain, although to a much lesser extent. It is composed of approximately 99% introns, and the coding region is represented in only 86 exons.
Three different isoforms of this protein are recognized that come from the translation of messengers that are transcribed from three different promoters: one that is only found in cortical and hippocampal neurons, another in Purkinje cells (also in the brain) , and the latter in muscle cells (skeletal and cardiac).
Since the dystrophin gene can be "read" from different internal promoters, there are different isoforms of this protein that are, of course, different sizes. Based on this, the structure of the "complete" and "short" isoforms is described below..
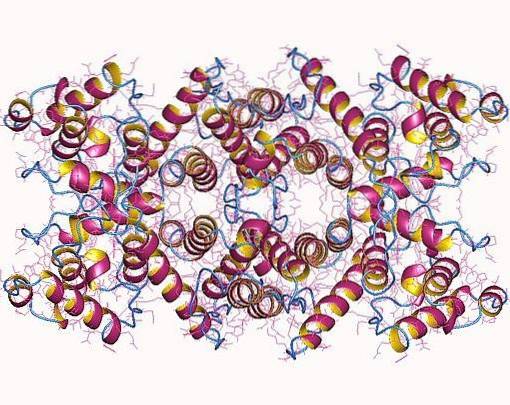
The "whole" isoforms of dystrophin are rod-shaped proteins that possess four essential domains (N-terminal, central domain, cysteine-rich domain, and C-terminal domain) that together weigh just over 420 kDa and are roughly 3,685 amino acid residues.
The N-terminal domain is similar to α-actinin (an actin-binding protein) and can be between 232 and 240 amino acids, depending on the isoform. The core or rod domain is composed of 25 spectrin-like triple helical repeats and has about 3,000 amino acid residues..
The C-terminal region of the central domain, which is made up of a domain rich in cysteine repeats, has about 280 residues and is very similar to the calcium-binding motif present in proteins such as calmodulin, α-actinin, and β. -spectrine. The C-terminal domain of the protein is made up of 420 amino acids.
Since the dystrophin gene has at least four internal promoters, there may be proteins with different lengths, which differ from each other due to the absence of any of their domains..
Each of the internal promoters has a unique first exon that separates into exons 30, 45, 56 and 63, generating products of 260 kDa (Dp260), 140 kDa (Dp140), 116 kDa (Dp116) and 71 kDa (Dp71 ), which are expressed in different regions of the body.
Dp260 is expressed in the retina and coexists with “full” brain and muscle forms. Dp140 is found in the brain, retina, and kidneys, while Dp116 is only found in adult peripheral nerves and Dp71 is found in most non-muscular tissues.
According to various authors, dystrophin has various functions that not only imply its participation as a protein of the cytoskeleton.
The main function of dystrophin, as a molecule associated with the membrane of nerve and muscle cells, is to interact with at least six different integral membrane proteins, with which it binds to form dystrophin-glycoprotein complexes..
The formation of this complex generates a “bridge” through the membrane of the muscle cells or sarcolemma and connects “flexibly” the basal lamina of the extracellular matrix with the internal cytoskeleton.
The dystrophin-glycoprotein complex functions in membrane stabilization and in the protection of muscle fibers against necrosis or damage caused by contraction induced for long periods of time, which has been demonstrated through reverse genetics..
This "stabilization" is often seen as analogous to what a similar protein known as spectrin provides cells such as erythrocytes circulating in the blood when they pass through narrow capillaries..
Dystrophin or, rather, the protein complex that it forms with the glycoproteins in the membrane not only has structural functions, but it has also been pointed out that it may have some functions in cell signaling and communication.
Its location suggests that it may participate in the transmission of tension from actin filaments in the sarcomeres of muscle fibers through the plasma membrane to the extracellular matrix, since this is physically associated with these filaments and with the extracellular space..
Evidence of other functions in signal transduction has emerged from some studies carried out with mutants for the dystrophin gene, in which defects in the signaling cascades that have to do with programmed cell death or cell defense are observed..


Yet No Comments