
The Joubert syndrome is a disorder of genetic origin that is characterized by a decrease in muscle tone, coordination problems, abnormal eye movements, altered breathing patterns and intellectual disability (Joubert Syndrome Foundation, 2016).
All these alterations are due to an autosomal genetic transmission that will cause important brain abnormalities, reduction of the cerebellar vermis, as well as abnormalities in the structure of the brain stem (National Institute of Neurological Disorders and Stroke, 2016).
In addition, Joubert syndrome is part of a group of disorders called ciliopathies that involve a dysfunction of a part of the cells called cilia. Joubert Syndrome Foundation, 2016).
The initial description of this pathology was made by Marie Joubert and collaborators in 1968, in which four cases were described. In the patients there was partial or total absence of the cerebellar vermis, neonatal episodic ampnea-hypernea syndrome, abnormal eye movements, ataxia and mental retardation (Angemi and Zucotti, 2012).
In addition, this syndrome was also associated with different multiorgan alterations, such as liver fibrosis, polydactyly, nephronoptysis or retinal dystrophy (Angemi and Zucotti, 2012).
In terms of treatment, there is currently no cure for Joubert syndrome. Therapeutic interventions are aimed at symptomatic control and support, children's physical and intellectual stimulation and occupational therapy (National Institute of Neurological Disorders and Stroke, 2016).
Article index
Joubert syndrome (JS) is a type of pathology of genetic origin that is characterized by a congenital malformation in the areas of the brain stem and an agenesis (partial or complete absence) or hypoplasia (incomplete development) of the cerebellar vermis, which can cause (Ophatnet, 2009).
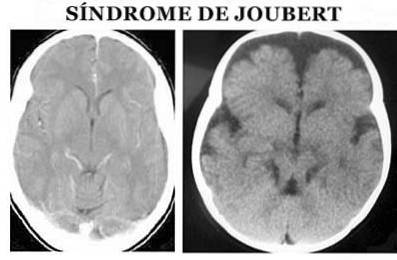
More specifically, at the anatomical level it is characterized by the so-called molar sign of the midbrain: agenesis or hypoplasia of the cerebellar vermis, narrowing of the superior cerebellar peduncles with thickening, elongation and lack of decussation and deep interpeduncular fossa (Angemi and Zuccoti, 2012).
It is a disorder that can affect many areas and organs of the body, so the signs and symptoms vary considerably among affected people (U.S. National Library of Medicine, 2011).
Most of those affected suffer from weakened muscle tone (hypotonia) and motor coordination difficulties (Ataxia). Other characteristic features are: episodes of altered breathing, nystagmus (involuntary and arrhythmic movement of the eyes), delayed motor development and variable intellectual difficulties (U.S. National Library of Medicine, 2011).
The prevalence of Joubert syndrome has been estimated to be approximately 1 / 80,000 to 1 / 100,000 cases of live births. Worldwide, more than 200 clinical cases have been registered (Angemi and Zuccoti, 2012).
Many specialists consider that these figures are underestimated, since Joubert syndrome has a wide range of affectations and is widely underdiagnosed (U.S. National Library of Medicine, 2011).
Much of the clinical symptoms of Joubert syndrome are more than evident in childhood, many affected children have significant motor delays (National Organization for Rare Disease, 2011).
The most common characteristics of the clinical course are: lack of muscle control (ataxia), altered breathing patterns (hypercapnia), sleep apnea, abnormal eye movements (nystagmus) and low muscle tone (National Organization for Rare Disease, 2011).
On the other hand, some of the alterations that may be associated with Joubert syndrome include: altered development of the retina, abnormalities in the iris, strabismus, kidney and / or liver alterations, protrusion of the membranes that cover the brain, among others ( National Organization for Rare Disease, 2011).
All the alterations derived from this syndrome are encompassed in several areas: neurological, ocular, renal, and musculoskeletal alterations (Bracanti et al., 2010).
The most characteristic neurological alterations of Joubert syndrome are Bracanti et al., 2010): hypotonia, ataxia, generalized delay in development, intellectual alterations, alteration of respiratory patterns and abnormal eye movements.
On a physical level, the retina is one of the organs affected by Joubert syndrome. The alterations in this organ appear in the form of retinal dystrophy, due to a progressive degeneration of the cells responsible for photo reception..
Clinically, ocular alterations can range from congenital retinal blindness to progressive retinal degeneration.
On the other hand, it is also possible to observe the presence of the coloboma. This ocular alteration is a congenital defect that affects the ocular iris and appears as a hole or slit.
Pathologies related to kidney function affect more than 25% of those affected by Joubert syndrome.
In many cases, kidney abnormalities can remain asymptomatic for several years or begin to manifest with nonspecific signs, until it presents as acute or chronic renal failure.
From the first descriptions of this pathology, a frequent clinical finding is polydactialia (genetic disorder that increases the number of fingers or toes).
In addition, it is also common to observe orofacial or structural anomalies at the level of the spine.
Experimental studies have classified Joubert syndrome as an autosomal recessive disorder (National Organization for Rare Disease, 2011).
An autosomal recessive genetic disorder means that two copies of an abnormal gene must be present for the trait or disease to present (National Institutes of Health, 2014).
Therefore, a recessive genetic alteration occurs when a person inherits the same abnormal gene for the same trait from each parent. If an individual only receives one copy of the gene related to the disease, they will be a carrier but will not show symptoms (National Organization for Rare Disease, 2011).
In addition, at least ten genes have been identified as one of the possible causes of Joubert syndrome (National Organization for Rare Disease, 2011).
A mutation in the AHI1 gene is responsible for this pathological condition in approximately 11% of affected families. In people who have this genetic alteration, vision alterations are common due to the development of retinal dystrophy (National Organization for Rare Disease, 2011).
The nphp1 gene mutation is the cause of approximately 1-2% of Joubert syndrome cases. In individuals with this genetic alteration, kidney alterations are common (National Organization for Rare Disease, 2011).
On the other hand, a CEP290 gene mutation is the cause of 4-10% of Joubert syndrome cases (National Organization for Rare Disease, 2011).
In addition, mutations in the genes TME67, JBTS1, JBTS2, JBTS7, JBTS8 and JBTS9 are also related to the development of Joubert syndrome (National Organization for Rare Disease, 2011).
The diagnosis of Joubert syndrome is made on the basis of physical symptoms. It is necessary to perform both a detailed physical examination, as well as the use of different diagnostic tests, especially magnetic resonance images (Ophatnet, 2009).
In addition, molecular genetic tests are also often used to identify genetic alterations that have been demonstrated in 40% of cases of Joubert syndrome (National Organization for Rare Disease, 2011).
On the other hand, it is also possible to make a prenatal diagnosis of this pathology through fetal ultrasound and molecular analysis, especially in families with a genetic history of Joubert syndrome (Ophatnet, 2009).
When the most characteristic features of Joubert syndrome occur in combination with one or more additional physical pathologies, a diagnosis of Joubert syndrome and related disorders (JSRD) may be made (U.S. National Library of Medicine, 2011).
Therefore, depending on the type of related pathology associated with the presence of Joubert syndrome, we can find subtypes of this. However, the Joubert syndrome classification system is still in an evolutionary phase due to the discovery of genetic contributions and the greater understanding of phenotypic correlations..
We can therefore find (Bracanti et al., 2010):
The treatment used in Joubert syndrome is symptomatic and supportive of the underlying pathologies. In addition to pharmacological interventions, it is common to use early physical and cognitive stimulation (National Institute of Neurological Disorders and Stoke, 2016).
When respiratory alterations are significant, especially in the initial phases of life, it is necessary to monitor respiratory function (National Institute of Neurological Disorders and Stoke, 2016).
On the other hand, the identification and control of ocular degeneration, kidney complications, and the rest of the complications related to Joubert syndrome, should be carried out as early as possible to adjust the therapeutic measures (National Institute of Neurological Disorders and Stoke, 2016).



Yet No Comments