
The Williams syndrome it is a developmental disorder of genetic origin that is associated with a characteristic profile of physical and cognitive impairments. Specifically, at the clinical level, it is characterized by 4 cardinal points: 1) atypical facial features and characteristics, 2) generalized delay in psychomotor development and specific neurocognitive profile, 3) cardiovascular alterations and t) the possibility of developing infantile hypercalcemia.
Despite the fact that Williams syndrome is considered a rare disease, there are thousands of affected people around the world. Regarding the diagnosis, the clinical examination usually provides the necessary findings for its establishment, however, to rule out other pathologies and false positives, a genetic study is usually launched through various techniques.
On the other hand, there is neither a cure for Williams syndrome nor a standard treatment protocol, so most of the therapeutic interventions will try to regulate medical complications. In addition, it will be essential to include early care programs, individualized special education and neuropsychological stimulation in the interventions..
Article index
Williams syndrome is a developmental disorder that can significantly affect different areas.
Generally, this pathology is characterized by the presence of atypical facial features or cardiovascular alterations, moderate intellectual disability, learning problems and distinctive personality traits..
Thus, the first patient with Williams syndrome was described by Dr. Guido Fanconi, in a 1952 clinical report. However, it was the cardiologist Joseph Williams who in 1961 precisely identified this pathology, at the same time that it was described by the German Beuren.
Due to this, Williams syndrome receives its name from both authors (Williams-Beuren syndrome), or simply from the first.
Despite the fact that, until a few years ago, the identification of the pathology was carried out based on the phenotypic characteristics, in 1993 Edward et al. Found a genetic abnormality in chromosome 7q 11.23 as the etiological cause..
Although Williams syndrome is associated with a wide variety of secondary medical complications, it does not have a high mortality rate. In many cases, affected individuals are capable of reaching an independent functional level..
Williams syndrome is considered a rare or rare genetic disorder.
The Williams Syndrome Association, among other institutions, have estimated that Williams syndrome has a prevalence of approximately 1 case per 10,000 people worldwide. Specifically, it has been identified that in the United States there may be around 20,000 or 30,000 affected.
Regarding the distribution of the pathology by sex, there are no recent data that indicate a higher prevalence in any of them, in addition, no differences have been identified between geographic regions or ethnic groups.
On the other hand, we also know that Williams syndrome is a sporadic medical condition, although some cases of family transmission have been described.
Williams syndrome, like other pathologies of genetic origin, has a clinical course characterized by multisystem involvement.
Many authors, such as González Fernández and Uyaguari Quezada, describe the clinical spectrum of Williams syndrome categorized into several areas: biomedical characteristics, psychomotor and cognitive characteristics, psychological and behavioral characteristics, among others..
The physical affectation present in Wiliams syndrome is diverse, among the most frequent clinical findings we can observe:
Delayed or slowed development can already be detected during pregnancy. Children affected by Williams syndrome are often born with low weight and height. In addition, once the adult stage is reached, the total height is usually lower than that of the general population, approximately 10-15 cm.
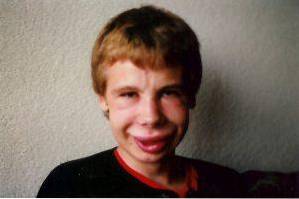
Facial alterations are one of the most characteristic clinical findings in this syndrome. In affected individuals we can observe a significantly narrow forehead, marked skin folds in the palpebral fissure, strabismus, stellate iris, short and flattened nose, prominent cheekbones and a smaller chin than usual..
In the case of alterations related to the development of muscles and bones, it is possible to observe the presence of reduced muscle tone and strength, joint laxity, scoliosis, contractures, among others. Visually, a posture characterized by drooping shoulders and semi-flexed lower extremities can be observed..
Although no significant abnormalities or malformations are usually found in the auditory pinna, in all cases an increase in auditory sensitivity develops. Affected individuals tend to perceive or experience certain sounds as annoying or painful.
The skin tends to have little elasticity, so it is possible to observe early signs of aging. In addition, hernias may develop, especially in the inguinal and umbilical region..
The different abnormalities in the heart and blood vessels constitute the most significant medical complication, since they can endanger the survival of the affected person.
Among the cardiovascular anomalies, some of the most common are supravalvular aortic stenosis, pulmonary branch stenosis, aortic valve stenosis. All these alterations, at a clinical level, can affect other vascular territories and even the brain, due to the development of arterial hypertension.
Abnormalities related to kidney and bladder function are very common. In addition, an accumulation of calcium (nephrocalcinosis), urinary urgency or nocturnal enuresis can also be detected.
At the cognitive level, the most significant characteristics are constituted by a generalized delay in the acquisition of motor skills, moderate intellectual delay and various alterations related to visual perception.
Various alterations related to balance and coordination problems are described, which are mainly due to the presence of musculoskeletal abnormalities and which will cause, among other things, a delay in gait acquisition, final motor skills, etc..
It is possible to find a moderate mental retardation, the typical IQ of those affected usually oscillates between 60 and 70. Regarding the specific areas that are affected, there is a clear asymmetry: in addition to psychomotor coordination, perception and visual integration, it usually be clearly affected, while areas such as language are usually more developed.
In the most initial stages, there is usually a delay in the acquisition of language skills, however, it usually recovers around 3-4 years. Children with Williams syndrome tend to have good expressive communication, are able to use contextualized vocabulary, correct grammar, eye contact, facial expressions, etc..
One of the most significant findings in Williams syndrome is the exceptional social behavior of those affected. Although in some cases anxiety crises or excessive worries may occur, they are very empathetic and sensitive.
The most recent research has indicated that the cause of Williams syndrome is found in various genetic alterations on chromosome 7. Chromosomes carry the genetic information of each person and are located in the nucleus of the body's cells.
In humans, we can find 46 chromosomes that are distributed in pairs. These are numbered from 1 to 23, except for the last pair made up of the sex chromosomes, called XX in the case of women, XY in the case of men. Thus, within each chromosome there can be an infinity of genes.
Specifically, the abnormal process identified in Williams syndrome is a microcelection or breakdown of a DNA molecule that confirms this chromosome. Normally, this type of error takes place in the development phase of the male or female gametes..
Genetic abnormalities are found in the 7q11.23 area, in which more than 25 different genes related to the characteristic clinical pattern of this pathology have been identified..
Some of the genes, such as Clip2, ELN, GTF21, GTF2IRD1 or LIMK1, are absent in those affected. Loss of ELN is related to connective tissue, skin and cardiovascular abnormalities.
On the other hand, some research indicates that the loss of the Clip2, GTF2I, GTF2IRD1 and LIMK1 genes may explain alterations in visuoperceptive processes, behavioral phenotype or cognitive deficits.
Furthermore, specifically, the GTF2IRD1 gene seems to play a prominent role in the development of atypical facial features. For its part, the NCF1 gene seems to be related to a high risk of developing Hypertension.
Until recent years, the diagnosis of Williams syndrome was made exclusively based on the observation of phenotypic characteristics (facial alterations, intellectual disability, specific cognitive deficits, among others).
However, currently, the diagnosis of Williams syndrome is usually made in two stages: analysis of clinical findings and confirmatory genetic studies. Thus, the clinical diagnosis usually includes:
- Physical and neurological examination and evaluation.
- Analysis of growth parameters.
- Cardiorespiratory system examination.
- Nephrourological examination.
- Analysis of calcium levels in urine and blood.
- Ophthalmological analysis.
On the other hand, genetic analysis is used to confirm the presence of genetic alterations compatible with Williams syndrome, among the most common tests is the fluorescent in situ hybridization (FIHS) technique..
After the extraction of a blood sample, the in situ hybridization technique is performed by marking DNA probes that are detected under a fluorescent light..
There is no specific treatment for Williams syndrome, however, this pathology is associated with multiple complications in different organs, so medical interventions will be oriented towards the treatment of these.
The authors González Fernández and Uyaguari Quezada emphasize that all interventions must have a marked multidisciplinary nature, allowing for the treatment of the symptomatic variety characteristic of this syndrome. In addition, they also point out various therapeutic measures depending on the affected area:
In this case, medical complications such as cardiac alterations or musculoskeletal malformations usually require treatment based mainly on the administration of drugs and surgical procedures. Medical professionals from different areas (pediatricians, cardiologists, ophthalmologists, etc.) usually participate in the treatment of physical symptoms..
Cognitive deficits such as visual-perceptual alteration or linguistic delay should be addressed from the early stages. Cognitive stimulation and rehabilitation will be a determining factor in achieving an autonomous life during adulthood.
Although those affected by Williams syndrome usually present good social functioning, on some occasions they tend to show excessively anxious behaviors and develop persevering behaviors or phobias.
Therefore, in these cases it will be essential to implement a psychological approach, through various strategies that are effective to minimize these problems or difficulties..



Yet No Comments